
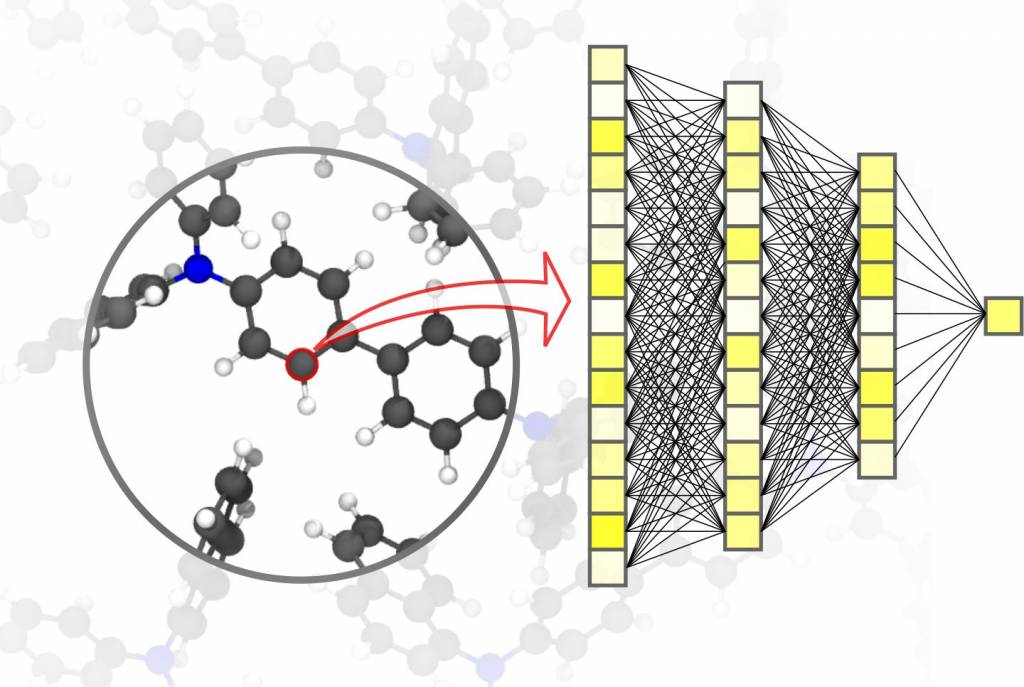
Erforschung, Entwicklung und Herstellung neuer Materialien hängen von schnellen und genauen Simulationsmethoden ab. Methoden der KI und des maschinellen Lernens können Materialsimulationen entscheidend voranbringen. „Schnellere Simulationssysteme ermöglichen es, größere und komplexere Materialsysteme rein virtuell zu entwickeln, sie bis auf die atomare Ebene hinunter zu verstehen und zu optimieren,“ erklärt Professor Pascal Friederich, Leiter der Forschungsgruppe AiMat – Artificial Intelligence for Materials Sciences am Institut für Theoretische Informatik (ITI) des KIT.
Hohe Präzision vom Atom bis zum Werkstoff
Algorithmen für maschinelles Lernen ermöglichen künstlicher Intelligenz, die eingegebenen Daten nicht nur zu verarbeiten, sondern in großen Datensätzen Muster und Korrelationen zu finden, sowie selbstständig Vorhersagen und Entscheidungen zu treffen. Bei Materialsimulationen kommt es darauf an, eine hohe Präzision über verschiedene Zeit- und Größenskalen – vom Atom bis zum Werkstoff – zu erreichen und zugleich die Rechenkosten zu begrenzen. Aktuelle Anwendungen sind z.B. kleine organische Moleküle und große Biomoleküle, strukturell ungeordnete feste, flüssige und gasförmige Materialien sowie komplexe kristalline Systeme.
Noch mehr Tempo mit hybriden Methoden
Um die Möglichkeiten der Materialsimulationen zukünftig noch zu erweitern, schlagen die Forschenden vor, hybride Methoden zu entwickeln: Diese verbinden Verfahren des maschinellen Lernens (ML) und der molekularen Mechanik (mm) miteinander. Solche hybriden Methoden könnten künftig z.B. die Simulation großer Biomoleküle oder enzymatischer Reaktion noch einmal deutlich beschleunigen.